

[ad_1]
Stylized picture of a CLASSIX clustering end result overlaid on prime of a coronavirus illustration. Credit score: College of Manchester, CDC / Alissa Eckert, MSMI; Dan Higgins, MAMS
An AI framework aids in figuring out and monitoring new COVID-19 variants, utilizing a novel algorithm named CLASSIX to effectively course of giant genomic datasets and improve early detection efforts.
Scientists at The Universities of Manchester and Oxford have developed an AI framework that may determine and observe new and regarding COVID-19 variants and will assist with different infections sooner or later.
The framework combines dimension discount methods and a brand new explainable clustering algorithm referred to as CLASSIX, developed by mathematicians at The College of Manchester. This permits the short identification of teams of viral genomes which may current a threat sooner or later from large volumes of information.
The research, offered this week within the journal PNAS, may help conventional strategies of monitoring viral evolution, equivalent to phylogenetic evaluation, which at the moment require intensive handbook curation.
Roberto Cahuantzi, a researcher at The College of Manchester and first and corresponding writer of the paper, stated: “Because the emergence of COVID-19, we have now seen a number of waves of recent variants, heightened transmissibility, evasion of immune responses, and elevated severity of sickness.
“Scientists are actually intensifying efforts to pinpoint these worrying new variants, equivalent to alpha, delta, and omicron, on the earliest levels of their emergence. If we will discover a means to do that shortly and effectively, it is going to allow us to be extra proactive in our response, equivalent to tailor-made vaccine growth and will even allow us to remove the variants earlier than they turn out to be established.”
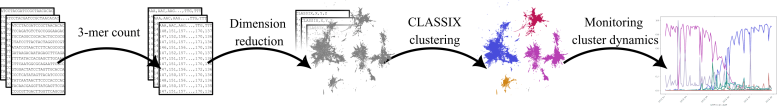
Diagram displaying the steps of the proposed methodology to determine emergent COVID-19 variants. Credit score: The College of Manchester
Like many different RNA viruses, COVID-19 has a excessive mutation price and quick time between generations that means it evolves extraordinarily quickly. This implies figuring out new strains which are more likely to be problematic sooner or later requires appreciable effort.
At the moment, there are virtually 16 million sequences obtainable on the GISAID database (the World Initiative on Sharing All Influenza Knowledge), which offers entry to genomic knowledge of influenza viruses.
Mapping the evolution and historical past of all COVID-19 genomes from this knowledge is at the moment achieved utilizing extraordinarily giant quantities of laptop and human time.
The described methodology permits the automation of such duties. The researchers processed 5.7 million high-coverage sequences in just one to 2 days on a typical fashionable laptop computer; this is able to not be doable for current strategies, placing the identification of regarding pathogen strains within the arms of extra researchers attributable to lowered useful resource wants.
Thomas Home, Professor of Mathematical Sciences at The College of Manchester, stated: “The unprecedented quantity of genetic knowledge generated through the pandemic calls for enhancements to our strategies to investigate it totally. The information is constant to develop quickly however with out displaying a profit to curating this knowledge, there’s a threat that it is going to be eliminated or deleted.
“We all know that human skilled time is restricted, so our strategy mustn’t change the work of people altogether however work alongside them to allow the job to be achieved a lot faster and free our consultants for different important developments.”
The proposed methodology works by breaking down genetic sequences of the COVID-19 virus into smaller “phrases” (referred to as 3-mers) represented as numbers by counting them. Then, it teams related sequences collectively based mostly on their phrase patterns utilizing machine studying methods.
Stefan Güttel, Professor of Utilized Arithmetic on the College of Manchester, stated: “The clustering algorithm CLASSIX we developed is far much less computationally demanding than conventional strategies and is absolutely explainable, that means that it offers textual and visible explanations of the computed clusters.”
Roberto Cahuantzi added: “Our evaluation serves as a proof of idea, demonstrating the potential use of machine studying strategies as an alert software for the early discovery of rising main variants with out counting on the necessity to generate phylogenies.
“While phylogenetics stays the ‘gold normal’ for understanding the viral ancestry, these machine studying strategies can accommodate a number of orders of magnitude extra sequences than the present phylogenetic strategies and at a low computational price.”
Reference: “Unsupervised identification of serious lineages of SARS-CoV-2 by means of scalable machine studying strategies” by Roberto Cahuantzi, Katrina A. Lythgoe, Ian Corridor, Lorenzo Pellis and Thomas Home, 13 March 2024, Proceedings of the Nationwide Academy of Sciences.
DOI: 10.1073/pnas.2317284121
[ad_2]
Supply hyperlink